SFB 1309

The Active Site of a Prototypical “Rigid” Drug Target is Marked by Extensive Conformational Dynamics
2020-09-23
Himanshu Singh, Chandan K. Das, Suresh K. Vasa, Kristof Grohe, Lars V. Schäfer, Rasmus Linser
Angew. Chem. Int. Ed., 2020, Volume 59, Issue 51, Pages 22916–22921
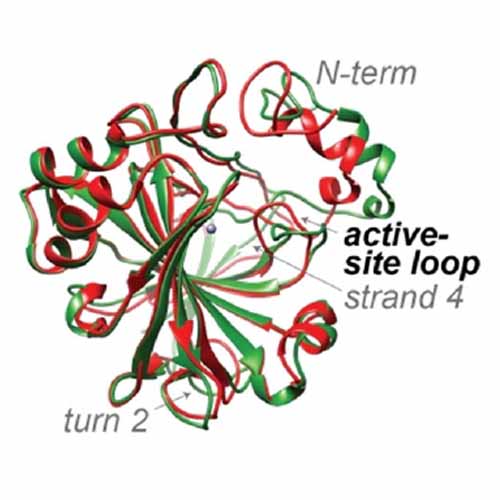
Drug discovery, in particular optimization of candidates using medicinal chemistry, is generally guided by structural biology. However, for optimizing binding kinetics, relevant for efficacy and off‐target effects, information on protein motion is important. Herein, we demonstrate for the prototypical textbook example of an allegedly “rigid protein” that substantial active‐site dynamics have generally remained unrecognized, despite thousands of medicinal‐chemistry studies on this model over decades. Comparing cryogenic X‐ray structures, solid‐state NMR on micro‐crystalline protein at room temperature, and solution NMR structure and dynamics, supported by MD simulations, we show that under physiologically relevant conditions the pocket is in fact shaped by pronounced open/close conformational‐exchange dynamics. The study, which is of general significance for pharmacological research, evinces a generic pitfall in drug discovery routines.