SFB 1309

Factors That Determine the Variation of Equilibrium and Kinetic Properties of QM/MM Enzyme Simulations: QM Region, Conformation, and Boundary Condition
2022-02-28
Darren Demapan, Jörg Kussmann, Christian Ochsenfeld, and Qiang Cui
J. Chem. Theory Comput., 18, 4, 2530–2542, 2022
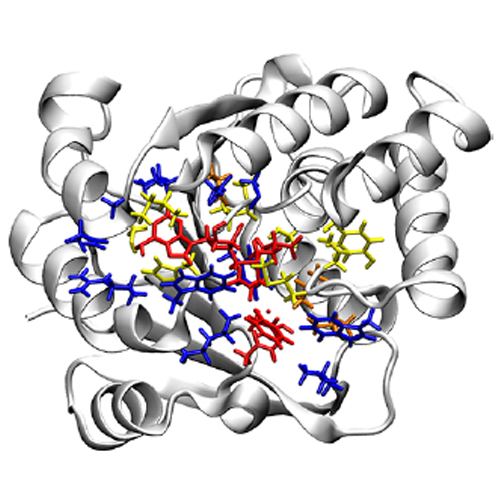
To analyze the impact of various technical details on the results of quantum mechanical (QM)/molecular mechanical (MM) enzyme simulations, including the QM region size, catechol-O-methyltransferase (COMT) is studied as a model system using an approximate QM/MM method (DFTB3/CHARMM). The results show that key equilibrium and kinetic properties for methyl transfer in COMT exhibit limited variations with respect to the size of the QM region, which ranges from ∼100 to ∼500 atoms in this study. With extensive sampling, local and global structural characteristics of the enzyme are largely conserved across the studied QM regions, while the nature of the transition state (e.g., secondary kinetic isotope effect) and reaction exergonicity are largely maintained. Deviations in the free energy profile with different QM region sizes are similar in magnitude to those observed with changes in other simulation protocols, such as different initial enzyme conformations and boundary conditions. Electronic structural properties, such as the covariance matrix of residual charge fluctuations, appear to exhibit rather long-range correlations, especially when the peptide backbone is included in the QM region; this observation holds when a range-separated DFT approach is used as the QM region, suggesting that delocalization error is unlikely the origin. Overall, the analyses suggest that multiple simulation details determine the results of QM/MM enzyme simulations with comparable contributions.