SFB 1309

Accelerating Hybrid Density Functional Theory Molecular Dynamics Simulations by Seminumerical Integration, Resolution-of-the-Identity Approximation, and Graphics Processing Units
2022-09-22
Henryk Laqua, Johannes C. B. Dietschreit, Jörg Kussmann and Christian Ochsenfeld
J. Chem. Theory Comput., 18, 10, 6010–6020, 2022
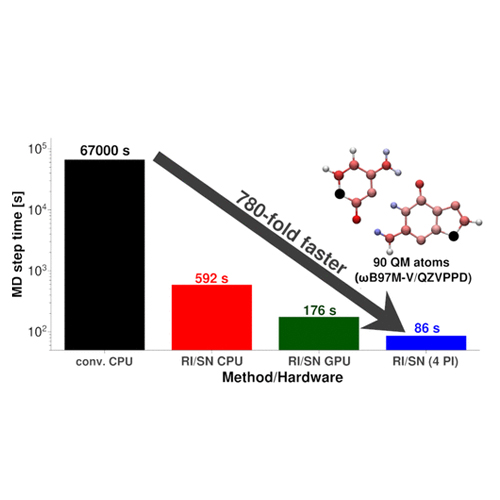
The computationally very demanding evaluation of the 4-center-2-electron (4c2e) integrals and their respective integral derivatives typically represents the major bottleneck within hybrid Kohn–Sham density functional theory molecular dynamics simulations. Building upon our previous works on seminumerical exact-exchange (sn-LinK) [Laqua, H., Thompsons, T. H., Kussmann, J., Ochsenfeld, C., J. Chem. Theory Comput.2020,16, 1465] and resolution-of-the-identity Coulomb (RI-J) [Kussmann, J., Laqua, H., Ochsenfeld, C., J. Chem. Theory Comput.2021,17, 1512], the expensive 4c2e integral evaluation can be avoided entirely, resulting in a highly efficient electronic structure theory method, allowing for fast ab initio molecular dynamics (AIMD) simulations even with large basis sets. Moreover, we propose to combine the final self-consistent field (SCF) step with the subsequent nuclear forces evaluation, providing the forces at virtually no additional cost after a converged SCF calculation, reducing the total runtime of an AIMD simulation by about another 25%. In addition, multiple independent MD trajectories can be computed concurrently on a single node, leading to a greatly increased utilization of the available hardware─especially when combined with graphics processing unit acceleration─improving the overall throughput by up to another 5 times in this way. With all of those optimizations combined, our proposed method provides nearly 3 orders of magnitude faster execution times than traditional 4c2e integral-based methods. To demonstrate the practical utility of the approach, quantum-mechanical/molecular-mechanical dynamics simulations on double-stranded DNA were performed, investigating the relative hydrogen bond strength between adenine–thymine and guanine–cytosine base pairs. In addition, this illustrative application also contains a general accuracy assessment of the introduced approximations (integration grids, resolution-of-the-identity) within AIMD simulations, serving as a protocol on how to apply these new methods to practical problems.