SFB 1309

Predicting 19F NMR Chemical Shifts: A Combined Computational and Experimental Study of a Trypanosomal Oxidoreductase–Inhibitor Complex
2020-04-02
Johannes C. B. Dietschreit, Annika Wagner, T. Anh Le, Philipp Klein, Hermann Schindelin, Till Opatz, Bernd Engels, Ute A. Hellmich, Christian Ochsenfeld
Angew. Chem. Int. Ed., 2020, 59, 12669–12673
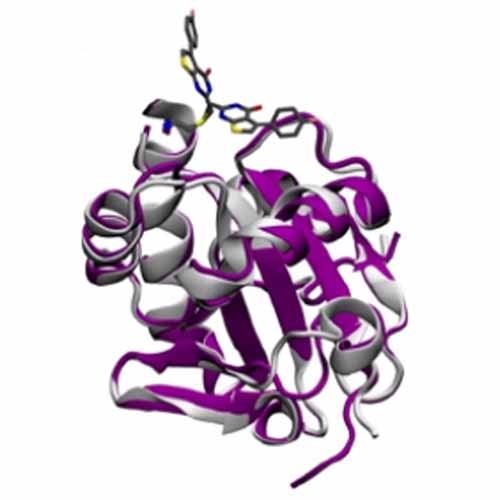
The absence of fluorine from most biomolecules renders it an excellent probe for NMR spectroscopy to monitor inhibitor–protein interactions. However, predicting the binding mode of a fluorinated ligand from a chemical shift (or vice versa) has been challenging due to the high electron density of the fluorine atom. Nonetheless, reliable 19F chemical‐shift predictions to deduce ligand‐binding modes hold great potential for in silico drug design. Herein, we present a systematic QM/MM study to predict the 19F NMR chemical shifts of a covalently bound fluorinated inhibitor to the essential oxidoreductase tryparedoxin (Tpx) from African trypanosomes, the causative agent of African sleeping sickness. We include many protein–inhibitor conformations as well as monomeric and dimeric inhibitor–protein complexes, thus rendering it the largest computational study on chemical shifts of 19F nuclei in a biological context to date. Our predicted shifts agree well with those obtained experimentally and pave the way for future work in this area.